家庭诊疗/儿童结缔组织或骨的遗传性疾病
| 医学电子书 >> 《默克家庭诊疗手册》 >> 儿童保健 >> 儿童骨骼肌肉系统疾病 >> 儿童结缔组织或骨的遗传性疾病 |
| 默克家庭诊疗手册 |
|
|
|
某些影响结缔组织的疾病是遗传性的。这些疾病包括埃-当氏综合征、马方综合征、弹性假黄瘤、皮肤松弛症和粘多糖病。骨软骨发育不良影响骨或软骨,骨硬化病影响骨骼。
埃-当氏综合征
埃-当氏(Ehlers-Danlos)综合征是一种非常罕见的遗传性结缔组织疾病,临床主要特征为关节过度灵活、皮肤弹性过强和组织脆弱。
埃-当氏综合征本身有几种变性,是由那些控制形成结缔组织的各种基因变异所致。多数患儿仅有十分灵活的关节(良性的运动过度),而无其他症状。经过一段时间后,患儿关节的过度灵活性可减轻。
目录 |
症状和诊断
皮肤可捏起10余cm,但是释放后能恢复到正常位置。关节可特别灵活。在身体的骨表面经常易形成广泛的伤痕,特别是在肘部、膝部和胫骨部。皮下可形成小而坚硬的圆形结节,这种结节可在X线片上显现。
轻度的碰撞可以引起宽阔的裂伤,并通常伴有少量出血。少数埃-当氏综合征患者有容易出血的倾向。缝线往往撕裂脆弱的结缔组织,因而增加了修补伤口的难度。身体内部器官也同样脆弱,也使手术变得困难。扭伤和脱位极为常见。患儿中,大略25%的逐渐出现脊柱异常弯曲(脊柱后侧凸)所致的驼背,并且90%的为扁平足。疝气和肠道憩室也极为常见。很少有胃肠道出血或破裂(穿孔)。
患此综合征的孕妇,可因其身体组织容易伸展而导致早产。如果胎儿已患此综合征,羊膜可能出现早破。此外由于结缔组织脆弱,采用剖腹产术或阴道口切开术(外阴切开术)来帮助婴儿娩出,可能会存在更多的困难。分娩前、分娩期或分娩后可能会发生严重出血。
预后和治疗
尽管患埃-当氏综合征会发生多种多样的并发症,但多数患者都具有正常的寿命。对于少数患者,并发症(如血管破裂)却会致命。
目前,尚无治愈该病的方法。由于组织的脆性,患者应避免受到外伤。经常穿用代保护性的衣服和垫子会有益处。如果埃-当氏综合征患者希望有小孩,应作遗传咨询,以确定婴儿可能被遗传的风险程度。
马方综合征
马方(Marfan)综合征是一种罕见的,导致眼、骨骼、心脏和血管异常的遗传性结缔组织疾病。
症状
马方综合征的遗传基因是显性的(见遗传学),但并非每一位遗传有此基因者均会患此病。患者的身高超过了他们的年龄和家庭成员。手臂间距(双臂从体侧平伸时手指间的距离)超过自身身高,手指细长。胸骨常有外凸或内陷畸形。关节极为灵活。扁平足和由于脊柱异常弯曲(脊柱后侧凸)的驼背,疝气等常见。通常,皮下脂肪少,上腭高。
眼晶体脱位。医生利用检眼镜,通过瞳孔即可看见晶体脱位的边缘。患者还可能有高度近视和视网膜(眼后的感光区)剥离。
可因主动脉壁层薄弱而逐渐引起重要的动脉扩张,形成动脉瘤(一种血管壁的凸出)(见主动脉瘤和夹层动脉瘤)。血液可渗入血管壁层之间(壁间动脉瘤),或动脉瘤破裂,导致大出血。随着主动脉扩张,导致心脏与主动脉交界处的主动脉瓣膜闭锁不全(动脉返流)(见心脏瓣膜疾病)。位于左心房和左心室间的二尖瓣,可能出现闭锁不全或脱垂(向后凸出进入左心房)。肺部可能出现充满积液的囊肿(见胸膜疾病)。囊肿可能破裂而引起气胸(空气进入胸膜腔),导致呼吸困难。
并发症的危险性主要取决于畸形的严重程度。主要的危险是主动脉突然破裂,导致迅速死亡。破裂很可能发生在运动期间。
诊断与治疗
如果一个异乎寻常地高、消瘦的人有上述典型症状的任何一种,医生可以怀疑其患马方综合征。然而,许多的该综合征患者可以不出现任何症状和异常。
治疗的主要目的是预防血管和眼睛的疾病。应每年检查一次眼睛。如果视力出现问题,应该立即就诊。
利血平或普萘洛尔可用来减轻血流对管壁施加的压力,预防主动脉扩张和破裂。如果主动脉已出现扩张,有时需要手术修补或置换受损部位。
患马方综合征的儿童身材往往趋向于长得很高。医生可以使用激素(雌激素和孕酮)来治疗长得太高的女孩。这种治疗通常可促使青春期在10岁时早熟,从而使其停止生长。
马方综合征患者若希望生育应该寻求遗传咨询,以确定他们的孩子被遗传马方综合征的危险程度。
弹力纤维性假黄瘤
弹力纤维性假黄瘤是一种遗传性结缔组织疾病,主要影响皮肤、眼睛和血管。
弹力纤维性假黄瘤主要影响弹性纤维。使颈部、腋下、腹股沟和脐周的皮肤增厚,起皱纹、无弹性和松弛,微黄的鹅卵石样花纹结节使皮肤像一种毛小鸡外观。视网膜的改变可能导致视力的严重损害,甚至失明。动脉可能变窄,导致胸痛和间歇性跛行(因为血液供给不足,导致活动时出现腿痛)。可能发生鼻出血和脑部、子宫及肠道的出血。高血压常见。
预后与疾病和动脉受损的严重程度有关。患者通常在30~70岁期间死于并发症。
皮肤松弛症
皮肤松弛症是一种罕有的皮肤极易延伸和松弛下垂的结缔组织性疾病。
皮肤松弛症主要影响弹性纤维。本症病因不清楚,通常是遗传性的,但常无家族史。有些病例遗传特征相当轻微,有些导致智力迟钝,而其他的一些可能是致命的。
症状
在遗传型,皮肤可能在出生时就非常松弛或出生后变得松弛。皮肤松弛在面部特别显著,所以患儿面容呈悲哀状或“丘吉尔”貌。典型的钩状鼻子。常有疝气和肠道憩室。肺气肿可能导致肺动脉高压症,出现轻微的肺心病。
非遗传型(获得型)皮肤松弛症的临床表现与遗传型有所不同。它发病较晚,并影响皮肤以外的其他部位,受累的皮肤看起来与遗传型皮肤不同,而且鼻子不是钩状的。特别在儿童或青少年,皮肤松弛症发病类似一些严重疾病,伴发烧、广泛的身体浆膜炎和严重的皮疹(多形性红斑)。在成人,疾病可能不知不觉地发生,有时因肺部并发症或主动脉破裂导致死亡。
诊断和治疗
对皮肤松弛症,通过检查皮肤极易确诊。
皮肤松弛症尚不能治愈。对遗传型患者,整形外科能够成功地改善外貌,但是,皮肤可能再次变松弛。整形外科对获得型的治疗少有成功。同样需要治疗肺部疾病,如肺气肿或肺动脉高压症。
粘多糖病
粘多糖病是一种以典型面部特征和骨骼、眼睛、肝脏以及脾脏畸形的遗传性疾病,有时伴随智力障碍。
这类疾病的根本原因是身体储存过多的粘多糖使体内粘多糖含量过高。粘多糖是结缔组织的主要成分。同样,过剩的粘多糖进入血液并且堆积于全身,部分随尿液排泄。粘多糖有许多不同的类型,需要不同的生化降解酶。所以,在许多粘多糖病的类型中,每一种特殊的遗传缺陷影响一种特殊的生化酶。
症状和诊断
通常,粘多糖病患儿在出生时没有症状,到婴儿期和儿童期,身材矮小、骨骼发育不良、多毛以及身体发育不良才逐渐明显。面容可能已经丑陋、厚嘴唇、张口以及鞍型鼻。依据粘多糖病的类型,受累的患儿有些有正常的智力,而多数患儿在出生时智力似乎正常,几年后却逐渐地显现出智力迟钝。少数类型的患儿,有眼睛受累,出现角膜浑浊和视力障碍。其家庭成员也可能有粘多糖病。
医生根据症状可以作出初步诊断。粘多糖病可以通过羊水或绒毛膜取样(见遗传疾病检查诊断),进行分离和筛查异常酶活性,在患儿出生前作出诊断。出生后,采集尿样品亦可筛查。但是,尿检查结果常常不准确,因此,必须进行血液和其他检查才能确定诊断。X线片上显示的骨骼畸形对诊断有帮助。
预后和治疗
预后依赖于粘多糖病的类型。某些类型可引起早期死亡。
目前,该病尚无治疗方法。置换异常酶的设想作用是有限的,或仅可获得暂时的成功。骨髓移植(见移植)对本病作用不大,而且,经常引起死亡或残疾。因此,对该病的治疗仍有争论。
骨软骨发育不良
骨软骨发育不良是一组骨或软骨生长异常的遗传性疾病。
许多类型的骨软骨发育不良均可引起身材矮小(侏儒)。软骨发育不良是各类肢体短小侏儒症中最常见的类型之一。该病发生率约在25000~40000活产婴儿中出现1例,男女和所有人种均可发病。
症状
每一种不同类型有不同的症状,例如,软骨发育不良的症状是肢体短小、膝内翻、大额、鞍形鼻以及驼背。
驼背
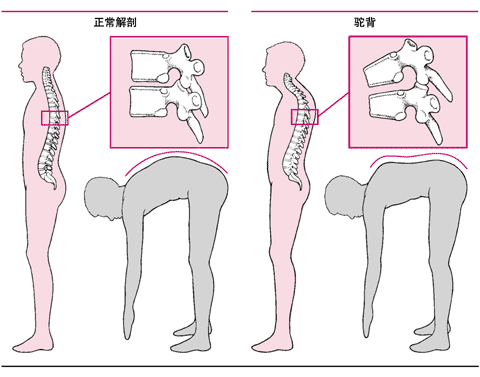
骨骼的疾病,如髋关节脱位是所有骨软骨发育不良的典型症状。在少数类型,第二椎骨的畸形(齿状突发育不良)使第二椎骨从第一椎骨脱位并且压迫脊髓,可立即导致死亡。
诊断和治疗
因为每一种类型由不同的遗传变异引起,其遗传性和病程不同,预后也不同,医生应尽力去获取准确的诊断。可在婴儿出生以前进行确诊。在一些病例可直接通过光纤镜(胎产镜)或超声扫描观察胎儿。假如一个骨软骨发育不良患者或其近亲希望要一个孩子,就得冒遗传因素影响孩子的危险。
当关节疾病严重影响患儿功能时,有时需施行人工关节置换术以替代这个受累关节。第二椎骨畸形必须给予手术固定,以预防脊髓损伤。
骨硬化病
骨硬化病(大理石样骨)是一种骨密度增加和骨骼成形异常的遗传性疾病。
骨硬化病在有些病人几乎不引起残疾,而另一些则为进行性的并且是致命的。一些类型没有明显的症状(迟发型);在另一些类型,症状在幼年即开始显现(早发型)。
大部分类型无特殊的治疗。必须外科手术缓解颅骨畸形所致的颅内高压。彻底切开骨周围神经,可以抑制骨骼的过度生长,如果这个过度生长的骨压迫神经,需要外科手术来减轻压迫。牙移植,可以预防特有的牙咬合异常(错合);牙正畸治疗可以纠正这些情况。骨的过度生长可能引起面部严重扭曲,有时会导致心理障碍。
迟发型
症状可出现在儿童期、少年和青年期。大理石样骨病(阿尔贝斯.舍恩贝格病)是迟发型中轻微的一种,可以没有症状。这种类型分布相当广泛,可出现在任何国家和任何人种中。通常,患儿在出生时骨骼正常,至儿童期骨密度增加。确诊常常带有偶然性,如在X线摄片中无意发现骨密度增高。患者面容、体格、智力和寿命正常,身体健康。有时,由于骨的过度生长压迫神经造成面瘫或耳聋。可逐渐出现轻微的贫血。
早发型
早发型骨硬化病较少见,但对婴儿却是一种潜在的致命性疾病。症状包括生长发育不良和体重不增,容易擦伤、异常出血以及贫血。肝脏和脾脏肿大,眼神经和面神经受损。患儿常在1岁内由于贫血、严重感染或出血而死亡。有些患儿X线片显示出骨密度增加,并随时间而加重。骨髓移植在有些患儿已获得成功,但是远期预后尚不清楚。
幼年变形性骨软骨病
骨软骨病是一组影响儿童发育期骨骺(出现于发育期不同的骨化中心)的疾病,结果为异常的骨生长和骨畸形。骨软骨病的病因尚不清楚。不同的疾病影响的骨骼不同,受侵袭的骨在股骨头骨软骨炎是股骨;在胫骨结节骨骺坏死症是胫骨;在脊柱骨软骨病是脊椎骨,以及在足舟骨骨软骨病是一种足的小骨(足舟骨)。
股骨头骨软骨炎
股骨头骨软骨炎(莱-卡-珀氏病)是股骨头的骨骺破坏。这种疾病在5~10岁的小孩中发生率为1/1000~1/5000,常见于男孩。疾病通常仅影响单侧髋关节,造成股骨头的供血不足,但是供血不足的原因不明。
主要症状是髋关节疼痛和行走困难。症状逐渐开始且进展缓慢。关节活动受限,大腿肌肉因缺乏活动而萎缩。在X线片上,股骨头开始变扁平;稍后,股骨头出现断裂。
治疗包括长期卧床休息、牵引、悬吊、石膏裤和夹板。一些专家建议使用整形外科来矫正髋关节的所有残余畸形。
该病不经治疗,可在2~3年后自愈。如果该病留有后遗症,在髋关节会增加并发骨关节炎的危险。如果疾病得到恰当治疗,则很少发生骨关节炎。
胫骨结节骨骺坏死症
胫骨结节骨骺坏死症(奥-施氏病)是在胫骨结节骨骺和软骨的炎症。
本病多发于10~15岁的男孩。该病的病因据认为是由于附着于胫骨粗隆下端的髌韧带过度牵拉所致的损伤。这个胫骨粗隆下端称为胫骨结节。
该病通常影响单侧胫骨。主要症状是在胫骨粗隆髌韧带附着部位的疼痛、肿胀和触痛。X线的膝关节片上可发现胫骨粗隆裂断。
唯一的治疗措施是用止痛剂减轻疼痛和限制有关的运动及过度的锻炼,尤其是避免膝关节过度弯曲。症状通常在病程几周或几月之后自行消失。有时候,必须用石膏模固定腿。在某些病例,可以局部注射氢化可的松,手术切除骨质碎片,骨钻孔引流减压直到康复,或者进行骨移植。
脊柱骨软骨病
脊柱骨软骨病是一种由于脊椎变型引起的背痛和驼背。
这种病多发于青少年,男孩较女孩多见。脊柱骨软骨病或许不是一种单一的疾病,而是具有相似特征的一组疾病。病因目前尚不清楚。
通常,轻型病例是在学龄儿童常规筛查脊柱畸形时才被检查出来。症状包括整个上背部的持续的轻度疼痛。脊柱上部的弯曲大于正常弯曲度。该病病程可持续一个较长时期,甚至达几年,但是症状轻微。症状消失后脊柱可能遗留轻微的弯曲。
轻度非进行性病例可通过减轻负重,减小背部张力,以及避免剧烈的活动来治疗。偶尔,驼背较严重时,必须采用脊柱支架或者睡硬板床。若是病情发展,有时候需用外科手术矫正。
足舟骨骨软骨炎
足舟骨骨软骨炎(克勒骨病)是一种罕见的足舟骨和软骨的炎症(骨软骨炎)。
该病常见于3~5岁的男孩。症状有患足肿胀、疼痛及触痛,尤其是在足弓的内侧。在足部负重或者行走时疼痛加重。常有跛行。该病病程往往为数月但是很少超过2年。止痛和避免足负重可减轻症状。倘若在早期阶段足底对足弓形进行恰当的支撑,膝下进行允许行走的石膏固定数周,对疾病可能有效。
| 关于“家庭诊疗/儿童结缔组织或骨的遗传性疾病”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |