生物化学与分子生物学/酶促反应的动力学
| 医学电子书 >> 《生物化学与分子生物学》 >> 酶 >> 酶促反应的动力学 |
| 生物化学与分子生物学 |
|
|
|
酶促反应动力学(kineticsof enzyme-catalyzed reactions)是研究酶促反应速度及其影响因素的科学。这些因素主要包括酶的浓度、底物的浓度、pH、温度、抑制剂和激活剂等。在研究某一因素对酶促反应速度的影响时,应该维持反应中其它因素不变,而只改变要研究的因素。但必须注意,酶促反应动力学中所指明的速度是反应的初速度,因为此时反应速度与酶的浓度呈正比关系,这样避免了反应产物以及其他因素的影响。
酶促反应动力学的研究有助于阐明酶的结构与功能的关系,也可为酶作用机理的研究提供数据;有助于寻找最有利的反应条件,以最大限度地发挥酶催化反应的高效率;有助于了解酶在代谢中的作用或某些药物作用的机理等,因此对它的研究具有重要的理论意义和实践意义。
目录 |
一、酶浓度对反应速度的影响
在一定的温度和pH条件下,当底物浓度大大超过酶的浓度时,酶的浓度与反应速度呈正比关系(图2-7)。
二、底物浓度对反应速度的影响
在酶的浓度不变的情况下,底物浓度对反应速度影响的作用呈现矩形双曲线(rectangular hyperbola)(图2-8)。
|
|
|
| 图2-7 酶浓度对反应初速度的影响 | 图2-8 底物浓度对反应初速度的影响 |
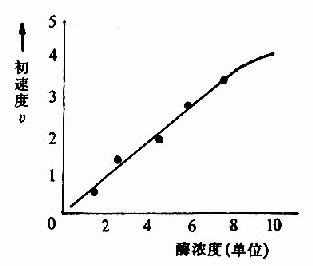
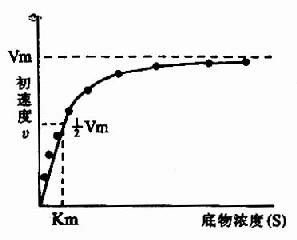

在底物浓度很低时,反应速度随底物浓度的增加而急骤加快,两者呈正比关系,表现为一级反应。随着底物浓度的升高,反应速度不再呈正比例加快,反应速度增加的幅度不断下降。如果继续加大底物浓度,反应速度不再增加,表现为0级反应。此时,无论底物浓度增加多大,反应速度也不再增加,说明酶已被底物所饱和。所有的酶都有饱和现象,只是达到饱和时所需底物浓度各不相同而已。
(一)米曼氏方程式
解释酶促反应中底物浓度和反应速度关系的最合理学说是中间产物学说。酶首先与底物结合生成酶椀孜锔春衔?中间产物),此复合物再分解为产物和游离的酶。
Michaelis和Menten在前人工作的基础上,经过大量的实验,1913年前后提出了反应速度和底物浓度关系的数学方程式,即著名的米椔戏匠淌?michaelismenten equation).
V=Vmax[S]/Km+[S]
Vmax指该酶促反应的最大速度,[S]为底物浓度,Km是米氏常数,V是在某一底物浓度时相应的反应速度。当底物浓度很低时,[S]《Km,则V≌Vmax/Km[S],反应速度与底物浓度呈正比。当底物浓度很高时,[S]》Km,此时V≌Vmax,反应速度达最大速度,底物浓度再增高也不影响反应速度(图2-9)。
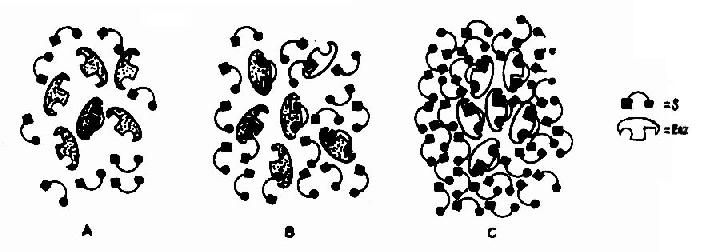
图2-9 酶与不同浓度的底物相互作用模式
(二)米-曼氏方程式的推导
米-曼氏方程式提出后又经riggs和Haldane的充实和发展,经补充和发展的米-曼氏方程工推导如下:
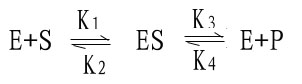 (1)
(1)
式中K1、K2、K3、K4分别为各向反应的速度常数。
从式(1)中知,ES的生成途径来自E+S和E+P,但其中E+P生成ES的速度极小(尤其在起始阶段,P的生成很少),可以忽略不计,又因为底物浓度大大超过酶的浓度,[S]》[E],中间产物ES中的S浓度可以忽略不计,因此,ES的生成速度为:
| d[ES] | = | K1([Et]-[ES]).[S] | (2) |
| dt |
其中[Et]-[ES]为游离酶的浓度,ES的分解速度为:
| - | [ES] | = | K2[ES]+K3[ES]=(K2+K3)[ES] | (3) |
| dt |
当反应体系处于稳态时,ES生成和分解的速度相等,即
K1([Et]-[ES]).[S]=(K2+K3)[ES]
| K2+K3 | = | [Et]-[ES] | .[S] |
| K1 | [ES] |
令K2+K3/K1=Km 则 Km=[Et]-[ES]/[ES].[S]
[ES]=[Et][S]/Km+[S] (4)
由于反应速度取决于产物P的生成量,故
V=K3[ES (5)
在酶促反应达最大速度时,所有的酶分子都已与底物结合形成中间产物,此时
[Et]=[ES] (6)
那么 Vmax=K3[Et] (7)
在(4)式两边乘以K3得:
K3.[ES]=K3.[Et][S]/Km+[S] 以(5)和(7)式代入,即:
V=Vmax[S]/Km+[S]
(三)米氏常数的意义
当反应速度为最大速度一半时,米氏方程可以变换如下:
½Vmax=Vmax[S]/Km+[S]
进一步整理可得到:
Km=[S]
可知,Km值等于酶反应速度为最大速度一半时的底物浓度。
因为Km=K2+K3/K1,当K2》K3,即ES解离成E和S的速度大大超过分离成E和P的速度时,K3可以忽略不计,此时Km值近似于ES解离常数KS,此时Km值可用来表示酶对底物的亲和力。
Km=K2/K1=[E][S]/[ES]=KS
Km值愈大,酶与底物的亲和力愈小;Km值愈小,酶与底物亲和力愈大。酶与底物亲和力大,表示不需要很高的底物浓度,便可容易地达到最大反应速度。但是KS值并非在所有酶促反应中都远小于K2,所以Ks值(又称酶促反应的底物常数)和Km值的涵义不同,不能互相代替使用。
Km值是酶的特征性常数,只与酶的性质,酶所催化的底物和酶促反应条件(如温度、pH、有无抑制剂等)有关,与酶的浓度无关。酶的种类不同,Km值不同,同一种酶与不同底物作用时,Km值也不同。各种酶的Km值范围很广,大致在10-1~10-6M之间。
当K3不远远小于K2和K1时,Km表示整个反应的化学平衡的常数。
如果Km值已知,任何底物浓度时酶的饱和度(形成中间产物的酶占总酶的比例,saturation fraction fEs)fEs便可计算出来。
fES=[ES]/[Et]=K3[ES]/K3[Et]=V/Vmax=[S]/Km+[S]
(四)Km和Vmax的求法
如图2?所示,底物浓度曲线是矩形双曲线。
从图中很难精确地测出Km和Vmax。为此人们将米氏方程进行种种变换,将曲线作图转变成直线作图。
1.双倒数作图(doublereciprocal plot or LineweaverBurk plot)
将米氏方程两边取倒数,可转化为下列形式:
1/V=Km/Vmax.1/[S]+1/Vmax
从图2-10可知,1/V对1/[S]的作图得一直线,其斜率是Km/V,在纵轴上的截距为1/Vmax,横轴上的截距为-1/Km。此作图除用来求Km和Vmax值外,在研究酶的抑制作用方面还有重要价值。
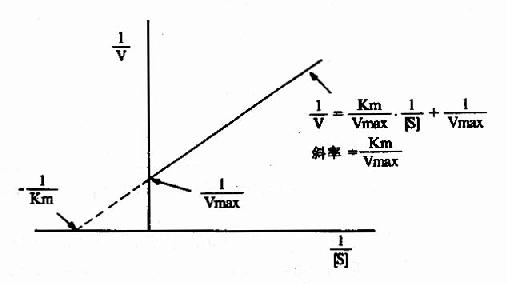
图2-10 双倒数作图法
![v对v/[s]作图法](http://p.ayxbk.com/images/f/ff/Gra28hdm.jpg)
图2-11 v对v/[s]作图法
2.V对V〖〗[S][SX)]法(EadieHofstee plot)
将米氏方程经移项整理后可写成
VKm+V[S]=Vm[S]
V[S]=Vm[S]-VKm
故V=Vm-KmV/[S]
以V为纵坐标对V/[S]横坐标作图,所得直线,其纵轴的截距为Vmax,斜率为Km(图2-11)。
必须指出米氏方程只适用于较为简单的酶作用过程,对于比较复杂的酶促反应过程,如多酶体系、多底物、多产物、多中间物等,还不能全面地籍此概括和说明,必须借助于复杂的计算过程。
三、pH对反应速度的影响
酶反应介质的pH可影响酶分子,特别是活性中心上必需基团的解离程度和催化基团中质子供体或质子受体所需的离子化状态,也可影响底物和辅酶的解离程度,从而影响酶与底物的结合。只有在特定的pH条件下,酶、底物和辅酶的解离情况,最适宜于它们互相结合,并发生催化作用,使酶促反应速度达最大值,这种pH值称为酶的最适pH(optimum pH)。它和酶的最稳定pH不一定相同,和体内环境的pH也未必相同。
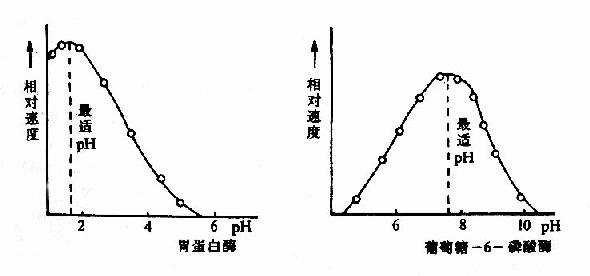
图2-12 胃蛋白酶和葡萄糖-6-磷酸酶的pH活性曲线
动物体内多数酶的最适pH值接近中性,但也有例外,如胃蛋白酶的最适pH约1.8,肝精氨酸酶最适pH约为9.8(见表2-2)。
表2-2 一些酶的最适pH
| 酶 | 最适pH | 酶 | 最适pH | 酶 | 最适pH |
| 胃蛋白酶 | 1.8 | 过氧化氢酶 | 7.6 | 延胡索酸酶 | 7.8 |
| 胰蛋白酶 | 7.7 | 精氨酸酶 | 9.8 | 核糖核酸酶 | 7.8 |
最适pH不是酶的特征性常数,它受底物浓度、缓冲液的种类和浓度以及酶的纯度等因素的影响。
溶液的pH值高于和低于最适pH时都会使酶的活性降低,远离最适pH值时甚至导致酶的变性失活。测定酶的活性时,应选用适宜的缓冲液,以保持酶活性的相对恒定。
四、温度对反应速度的影响
化学反应的速度随温度增高而加快。但酶是蛋白质,可随温度的升高而变性。在温度较低时,前一影响较大,反应速度随温度升高而加快,一般地说,温度每升高10℃,反应速度大约增加一倍。但温度超过一定数值后,酶受热变性的因素占优势,反应速度反而随温度上升而减缓,形成倒V形或倒U形曲线。在此曲线顶点所代表的温度,反应速度最大,称为酶的最适温度(optimum temperature)(图2-13)。
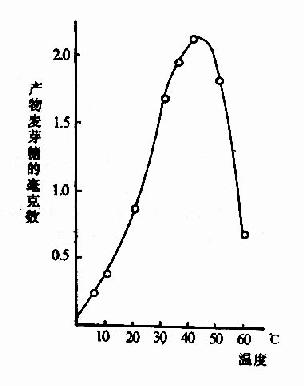
图2-13 温度对唾液淀粉酶活性影响
从动物组织提取的酶,其最适温度多在35℃~40℃之间,温度升高到60℃以上时,大多数酶开始变性,80℃以上,多数酶的变性不可逆。酶的活性虽然随温度的下降而降低,但低温一般不破坏酶。温度回升后,酶又恢复活性。临床上低温麻醉就是利用酶的这一性质以减慢组织细胞代谢速度,提高机体对氧和营养物质缺乏的耐受体,有利于进行手术治疗。
酶的最适温度不是酶的特征性常数,这是因为它与反应所需时间有关,不是一个固定的值。酶可以在短时间内耐受较高的温度,相反,延长反应时间,最适温度便降低。
五、抑制剂对反应速度的影响
凡能使酶的活性下降而不引起酶蛋白变性的物质称做酶的抑制剂(inhibitor)。使酶变性失活(称为酶的钝化)的因素如强酸、强碱等,不属于抑制剂。通常抑制作用分为可逆性抑制和不可逆性抑制两类。
(一)不可逆性抑制作用(irreversibleinhibition)
不可逆性抑制作用的抑制剂,通常以共价键方式与酶的必需基团进行不可逆结合而使酶丧失活性,按其作用特点,又有专一性及非专一性之分。
1.非专一性不可逆抑制
抑制剂与酶分子中一类或几类基团作用,不论是必需基团与否,皆可共价结合,由于其中必需基团也被抑制剂结合,从而导致酶的失活。某些重金属(Pb++、Cu++、Hg++)及对氯汞苯甲酸等,能与酶分子的巯基进行不可逆适合,许多以巯基作为必需基团的酶(通称巯基酶),会因此而遭受抑制,属于此种类型。用二巯基丙醇(british antilewisite,BAL)或二巯基丁二酸钠等含巯基的化合物可使酶复活。
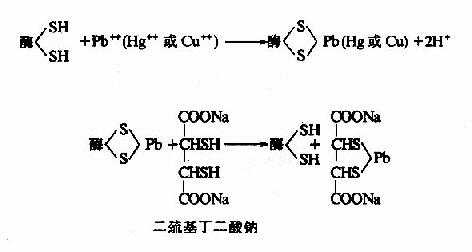
2.专一性不可逆抑制
此属抑制剂专一地作用于酶的活性中心或其必需基团,进行共价结合,从而抑制酶的活性。有机磷杀虫剂能专一作用于胆碱酯酶活性中心的丝氨酸残基,使其磷酰化而不可逆抑制酶的活性。当胆碱酯酶被有机磷杀虫剂抑制后,胆碱能神经末稍分泌的乙酰胆碱不能及时分解,过多的乙酰胆碱会导致胆碱能神经过度兴奋的症状。解磷定等药物可与有机磷杀虫剂结合,使酶和有机磷杀虫剂分离而复活。
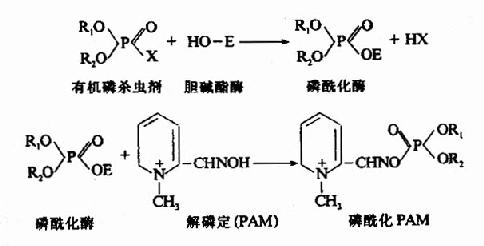
(二)可逆性抑制(reversible inhibition)
抑制剂与酶以非共价键结合,在用透析等物理方法除去抑制剂后,酶的活性能恢复,即抑制剂与酶的结合是可逆的。这类抑制剂大致可分为以下二类。
1.竞争性抑制(competitive inhibition)
(1)含义和反应式
抑制剂I和底物S对游离酶E的结合有竞争作用,互相排斥,已结合底物的ES复合体,不能再结合I。同样已结合抑制剂的EI复合体,不能再结合S。
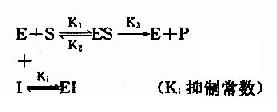
抑制剂I在化学结构上与底物S个相似,能与底物S竞争酶E分子活性中心的结合基团,因此,抑制作用大小取决于抑制剂与底物的浓度比,加大底物浓度,可使抑制作用减弱。
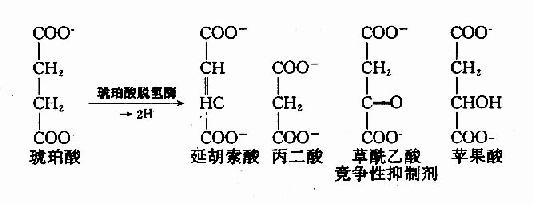
例如,丙二酸、苹果酸及草酰乙酸皆和琥珀酸的结构相似,是琥珀酸脱氢酶的竞争性抑制剂。
(2)反应速度公式及作图
按米氏公式推导方法,也可演算出竞争性抑制时,抑制剂、底物和反应速度之间的动力学关系及其双倒数方程式为:
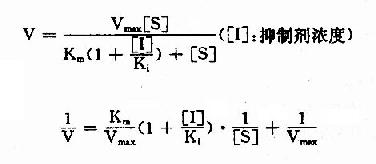
以1V分别为横坐标和纵坐标作图,此方程式可绘成竞争性抑制作用的特性曲线(图2-14)。
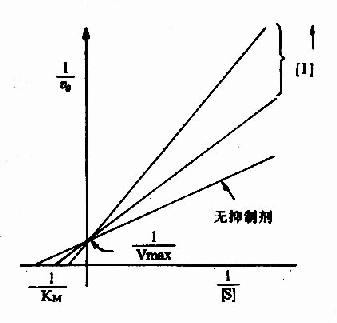
图 2-14 竞争性抑制
有竞争性抑制剂存在的曲线与无抑制剂的曲线相交于纵坐标I/Vmax处,但横坐标的截距,因竞争性抑制存在变小,说明该抑制作用,并不影响酶促反应的最大速度,而使Km值变大。
很多药物都是酶的竞争性抑制剂。例如磺胺药与对氨基苯甲酸具有类似的结构(如图2-15),而对氨基苯甲酸、二氢喋呤及谷氨酸是某些细菌合成二氢叶酸的原料,后者能转变为四氢叶酸,它是细菌合成核酸不可缺少的辅酶。由于磺胺药是二氢叶酸合成酶的竞争性抑制剂,进而减少菌体内四氢叶酸的合成,使核酸合成障碍,导致细菌死亡。抗菌增效剂-甲氧苄氨嘧啶(TMP)能特异地抑制细菌的二氢叶酸还原为四氢叶酸,故能增强磺胺药的作用。
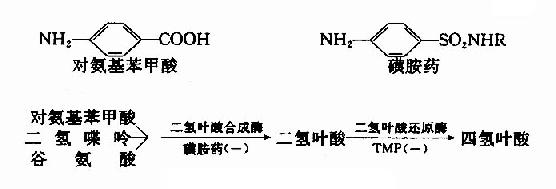
图2-15 磺胺药物的抑菌作用
2.非竞争性抑制(non-competitive inhibition)
(1)含义和反应式
抑制剂I和底物S与酶E的结合完全互不相关,既不排斥,也不促进结合,抑制剂I可以和酶E结合生成EI,也可以和ES复合物结合生成ESI。底物S和酶E结合成ES后,仍可与I结合生成ESI,但一旦形成ESI复合物,再不能释放形成产物P。
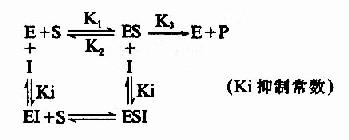
I和S在结构上一般无相似之处,I常与酶分子上结合基团以外的化学基团结合,这种结合并不影响底物和酶的结合,增加底物浓度并不能减少I对酶的抑制程度。
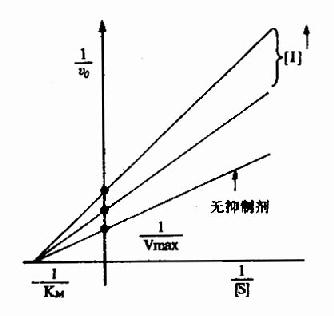
图2-16 非竞争性抑制
(2)反应速度公式及作图
按米氏公式推导方法可演算出非竞争性抑制时,抑制剂、底物浓度和反应速度之间动力学关系:
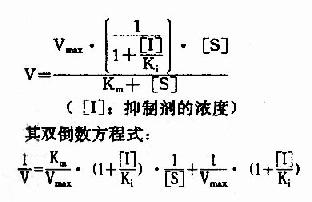
以1V分别为横坐标和纵坐标作图,此方程式可绘成非竞争性抑制作用的特性曲线(图2-16)。
有非竞争性抑制剂存在的曲线与无抑制剂存在的曲线相交于横坐标-1/Km处,纵坐标截距,因非竞争性抑制剂的存在而变大,说明该抑制作用,并不影响底物与酶的亲和力,而使酶促最大反应速度变小。
如赖氨酸是精氨酸酶的竞争性抑制剂,而中性氨基酸(如丙氨酸)则是非竞争性抑制剂。
总上所述,酶的竞争性和非竞争性抑制可通过双倒数作图加以区别。Vmax不因竞争性抑制剂的存在而改变,Km则不因非竞争性抑制剂的存在而改变。
六、激活剂对酶促反应速度的影响
能使酶活性提高的物质,都称为激活剂(activator),其中大部分是离子或简单的有机化合物。如Mg++是多种激酶和合成酶的激活剂,动物唾液中的α-淀粉酶则受Cl-的激活。
| 关于“生物化学与分子生物学/酶促反应的动力学”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |