医学遗传学/血红蛋白病的分类和分子基础
| 医学电子书 >> 《医学遗传学基础》 >> 单基因病 >> 基因突变致蛋白质合成异常 >> 血红蛋白病 >> 血红蛋白病的分类和分子基础 |
| 医学遗传学基础 |
|
|
|
1.异常血红蛋白病 异常血红蛋白(abnormalhemoglobin)是指由于珠蛋白基因突变导致珠蛋白肽链结构异常,如有临床表现者称为异常血红蛋白病或异常血红蛋白综合征。至今全世界已发现异常血红蛋白471种。国内已发现60种,其中20种是世界首报。尽管异常血红蛋白种类繁多,但仅约40%的异常血红蛋白对人体有不同程度的功能障碍。
(1)异常血红蛋白病的类型:
1)镰形细胞病(sickle cell disease):此病主要见于黑人。该病系由于β链第6位谷氨酸被缬氨酸取代,形成HbS,导致电荷改变,在脱氧情况下HbS聚合形成长棒状聚合物,使红细胞镰变,由于镰变引起血粘度增高,导致血管梗阻性继发症状,一过性剧痛(肌肉骨骼痛、腹痛),急性大面积组织损伤,心肌梗塞可致死,镰变细胞的变性降低还可引起溶血。HbS纯合子(HbSHbS)表现为镰形细胞性贫血,杂合子(HbAHbS)表现为镰形细胞性状,大部分无症状,但也可有轻度慢性贫血。
2)不稳定血红蛋白病(unstable hemoglobinpathies):已发现的不稳定血红蛋白在80种以上。由于Hb不稳定容易自发(或在氧化剂作用下)变性,形成变性珠蛋白小体(Heinz小体)。Heinz小体粘附红细胞膜上,导致了离子通透性增加;另外,由于变形性降低,当红细胞通过微循环时,红细胞被阻留破坏,导致血管内、外溶血。不稳定Hb病一般呈常染色体显性遗传(不完全显性),杂合子可有临床症状,纯合子可致死。临床表现与Hb不稳定程度、产生高铁血红蛋白的多少以及不稳定Hb的氧亲和力大小有关。轻者仅在服用磺胺等药物或有感染时溶血;重者需反复输血才能维持生命。
3)血红蛋白M病(HbM):HbM是因肽链中与血红素铁原子连接的组氨酸或邻近的氨基酸发生了替代,导致部分铁原子呈稳定的高铁状态,从而影响了正常的带氧功能,使组织供氧不足,导致临床上出现紫绀和继发性红细胞增多。本病呈常染色体显性遗传,杂合子HbM含量一般在30%以内,可引起紫绀症状。
4)氧亲和力改变的血红蛋白病:这类血红蛋白病是指由于肽链上氨基酸替代而使血红蛋白分子与氧的亲和力增高或降低,致运输氧功能改变。如引起Hb与氧亲和力增高,输送给组织的氧量减少,导致红细胞增多症;如引起Hb与氧亲和力降低,则使动脉血的氧饱和度下降,严重者可引起紫绀症状。
(2)异常血红蛋白的分子基础:异常血红蛋白的发生涉及基因突变的各种类型,概括举例如下。
1)单个碱基置换:大多数异常血红蛋白是由于珠蛋白基因发生单个碱基置换所致,其中多为错义突变。
①错义突变:例如镰形细胞贫血是β基因第6位密码子GAG变成GTG。中国人较常见的HbE是β基因第26位密码子由GAG(谷)→AAG(赖)所致。
②无义突变:例如HbMckees-Rock,其β链只有144个氨基酸组成,原因是β基因第145位酪氨酸密码子TAT改变为终止密码子TAA,使肽链合成提前终止。
③终止密码突变:例如Hb Constant Spring就是由于α珠蛋白基因第142位终止密码子TAA(mtRNA为UAA)突变为CAA(谷氨酰胺),结果α延长为172个氨基酸,这种突变基因转形成的mRNAI不稳定,所以导致α链合成减少,表现为α+地中海贫血。
2)移码突变:例如Hb Wagne是由于α链第138位丝氨酸密码子UCC丢失一个C,致使其3’端碱基顺序依次位移,重新编码,第142位终止信号变为可读密码,致使翻译至147位才终止(图4-13)。
3)整码突变:例如Hb Gum Hiu是β链缺失第91-95氨基酸(亮-组-半胱-门冬-赖),但其前后氨基酸顺序正常(图4-13)。

图4-13 人类α链和β链mRNAR不同突变类型
4)融合基因:例如Hb Lepore的类β链是由δ链和β链连接而成,肽链的N端像δ链,C端像β链,故称为δβ链,相反,Hb反Lepore(Hbanti-Lepore)其N端像β链,C端却像δ链,称为βδ链。这是由于染色体的错配联会和不等交换而形成的融合基因(fusion gene)(图4-14)。
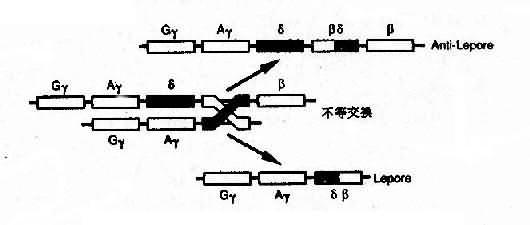
图4-14 血红蛋白融合基因形成机理
2.地中海贫血由于珠蛋白基因缺失或突变导致某种珠蛋的链合成障碍,造成α链和β链合成失去平衡面导致的溶血性贫血称为地中海贫血(thalassemia)。根据合成障碍的肽链不同可把地中海贫血分为α和β地中海贫血两类。此外还有少见的δβ和γβ地中海贫血。
(1)α地中海贫血(α-thalassemia,简称α地贫)是由于α珠蛋白基因的缺失或缺陷使α珠蛋白链(简称α链)的合成受到抑制而引起的溶血性贫血。如果一条16号染色体缺失1个α基因者称α+地贫(亦称α地2),缺失2个α基因者称为α0地贫(亦称α地1)。α地1的基因型可写作―/αα,α地2的单倍型则写成α-/αα.以上每种α地贫基因型与正常型配合可构成各种α地贫杂合子.各种α地贫基因型杂合子相互配合可构成各种纯合子或双重杂合子.α地中海贫血在我国多见于南方各省.
1)α地贫的临床类型:根据临床表现程度,依受累α基因数量不同而有差异,基本上可分为4类型.
①HbBart’s胎儿水肿综合征(HbBart’s hydrops fetalis syndrome):是两条16染色体的4个α基因全部缺失或缺陷,基因型为α0地贫纯合子(--/--),完全不能合成α链,不能形成胎儿HbF,相对过多的γ链形成γ四聚体(γ4)称为HbBart’s(γ4).HbBart’s对氧亲和力非常高,因而释放给组织的氧减少,造成组织严重缺氧导致胎儿水肿,引起死胎或新生儿死亡.患儿血红蛋白60%以上为HbBart’s,其余为 HbPortland.
患儿父母均为α0地贫杂合子,基因型为α/--.他们若再生育,则胎儿有1/4的机会为αHbBart’s水肿胎儿,1/4为正常人,1/2为α0地贫杂合子(α地1)
②血红蛋白H病:是α0地贫和α+地贫的双重杂合子,即有3个α基因缺失或缺陷,基因型为-α/--或α-/--,也可为ααT/--(αT代表有突变,如Hb Constant Spring)。因缺失3个α基因,只能合成少量α链,β链相对过多,形成β四聚体(β4),易被氧化,导致β4解体成游离的单链,游离β链沉淀聚积包涵体,附着于红细胞膜上,使红细胞膜受损,失去柔韧性,易被脾破坏,导致中等度或较严重的溶血性贫血,称为血红蛋白H病(Hbh disease) .
患者双亲的基因型多为α0地贫杂合子(αα/--)和α+地贫杂合子(α/αα),或为α0地贫杂合子(αα/--)和非缺失型地贫杂合子(ααT/αα)。婚配后其子女有1/4机会为正常人1/4为α+地贫杂合子,1/4为α0地贫杂合子及1/4为HbH病。如父母一方有αT,则导致非缺失型HbH病。
③轻型(标准型 )α地中海贫血:为α0地贫杂分子(--/αα)或α+地贫杂合子(α/α-),缺失两个α基因,间或有轻度贫血,我国主要是α0地贫杂合子。
轻型α地贫患者之间婚配,生育子女中可有1/4机会为HbBart水肿胎儿综合征。
④静止型α地中海贫血:仅缺失一个α基因,为α+地贫杂合子(-α/αα),无症状。
静止型α地贫与轻型地中海贫血个体婚配,可有1/4机会生育HbH病患儿。
2)α地中海贫血的分子基础:从基因缺陷程度来区分,可把α地贫分为缺失型和非缺失型(点突变)。
①基因缺失:可分为α+和α0地贫两种。α+地贫有两种基因型:左侧缺失(leftward deletion),缺失一个包括α2基因在内的DNA片段;②右侧缺失(right ward deletion), 缺失范围包括α2基因3’端和α1基因的5’端,结果形成了由α1的3’端和α2的5’端构成的融合基因。其发生机理是类α基因发生不等交换的结果。α0地贫基因缺失范围差别很大(图4-15)。

图4-15 类α基因簇缺失类型
②非缺失型(点突变:(类型见表4-1)。
(2)β地中海贫血:β地中海贫血(β梩halassemia,简称β地贫),是由于β珠蛋白基因的缺失或缺陷使β珠蛋白链(简称β链)的合成受到抑制而引起的溶血性贫血。完全不能合成β链者称β0地贫;能部分合成β链者(约为正常的5%-30%)称β+地贫。此外,还有δβ地贫。它们可以有不同的组合,即β0地贫纯合子(β0β0)、β0地贫双重杂合子(β0/β+)、β0地贫杂合子(β0βA)、β+地贫纯合子(β+/β+)和β+地贫杂合子(β+/βA)。β地贫在我国南方较常见。
1)临床分类:大致可有4种主要类型。
①重型β地中海贫血:患者是β+地贫、β0地贫或δβ0地贫的纯合子(其基因型分别为β+/β+、β0/β0和δβ0/δβ0)或是β+和β0地贫的双重杂合子(基因型为β0/β+)。这些患者的β链几乎不能合成,或合成量很少,以致无HbA或量很低,γ链的合成相对增加,使HbFt GbA2比率升高。由于HbF较HbA的氧亲和力高,在组织中不易释放出氧,所有β地贫患者有组织缺氧症状。组织缺氧促使红细胞生成素大量分泌,刺激骨髓的造血功能,使红骨髓大量增生,骨质受锓蚀致骨质疏松,可出现“地中海贫血面容”(头颅大,额顶及枕部隆起,鼻梁塌陷,上颌及牙齿前突,眼距宽,眼睑浮肿)。由于β链合成受抑制,过剩的游离α链形成α链包涵体,引起溶血性贫血,靠输血维持生命。
表4-1 点突变引起的地中海贫血
| 分子缺陷类型 |
| 1、生成无功能或稳定性降低的 ①无义突变 a116GAG---TAG,(G---T) ②移码突变 a130/31(--4bp) ③终止密码突变 142TAA---CAA(T---C) 形成Hb Constant Spring ④起始密码突变 a2ATG---ACG(T--C) 2、RNA加工突变 ①剪接改变 IVSI(GGTGAGGCT---GGCT) ②Poly(A)信号缺陷 AATAAAA ---AATAAG 3、产生不稳定H a2125CTG(亮)CCG(脯) 生成Hb Quong Sze |
②轻型β地中海贫血:患者是β+地贫、β0地贫或δβ0地贫的杂合子,基因型分别为β+/βA、β0/β+和δβ0/βA。这类患者由于还能合成相当量的β链,所以症状较轻,贫血不明显或轻度贫血。本病特点是HbA2升高(可达4%-8%)或(和)HbF升高。
③中间型β地中海贫血:患者通常是某些β地贫变异型的纯合子,如β+地贫(高F)/β+地贫(高F)或两种不同变异型地贫的双重杂合子,如β+,地贫/δβ+地贫。其症状介于重型和轻型之间,故称为中间型β地中海贫血。
④遗传胎儿血红蛋白持续增多症:患者是由于β基因簇中某些DNA片段的缺失或者点突变,使δ和β链合成受抑制,而γ链的合成明显增加,使成人红细胞内HbF含量持续增多,故称为遗传性胎儿血红蛋白持续增多症(hereditarypersistance of fetal hemoglobin,HPFH).HPFH的特点是HbF的成年人仍持续较高水平,无明显的临床症状。
2)β地中海贫血的分子基因:β地中海贫血迄今已发现100多种突变类型,其中10多种为缺失型,其余均为点突变。我国已报道17种点突变。
点突变:绝大多数β地中海贫血是由于β基因发生点突变所致,突变涉及基因内及旁侧表达顺序的各个环节。主要类型有4种。
a.编码区的无义突变、移码突变和起始密码突变:使生成的mRNA稳定性降低或形成无功能的mRNA,从而不能合成正常的β珠蛋白链,多数产生β0地贫,少数为β+地贫。例如无义突变密码子17(A→G)、43(G→T)都产生β0地贫;称码突变41/42(-TCTT),71/71(+A)和β0地贫;以及起始密码子突变ATG→AGG导致的β0都属这类。见于中国人。
b.非编码区IVS-1和IVS-2突变:影响前mRNA剪接等加工过程不能准确进行,形成异常的mRNA,导致β0或β+地贫。例如IVS-1的1位G→T为RNA拼接处改变;中国人中常见的IVS-2654,为内含子中由于碱基置换形成了一个的裂解信号,影响正常位点的剪接,产生异常mRNA;还有一种是内含子中剪接位点的通用顺序上的同义突变,从而激活内含子或外显子中隐蔽裂解位点(cryptic splicihng site,CSS)如IVS-15(G→C)。CSS即DNA的一段顺序在点突变后可以形成剪切识别顺序(CCTATTGGT)的第7个碱基G如变为A,则产生新的切点,即CCTATTAG↓T)。
c.影响转录的突变:这类突变主要集中于起始位点上游的启动子TATA框,使转录效率降低,mRNA生成量减少而产生β+地贫。中国人的-29A→G,-28A→G都属于这类。
d.RNA裂解部位缺陷:这类突变是由于异常的RNA加帽部位和多聚腺苷酸化信号的突变,从而影响RNA转录而不能准确裂解,产生不稳定的mRNA,使正常β链生成量减少,导致β+地贫。例如在mRNA加帽部位发生A→C颠换,引起β+地贫(亚洲人。)又如,多聚腺苷酸化信号AATAAA→AACAAA,引起β+地贫。
e.编码区的外显子突变引起剪接作用的改变:这类突变是由于编码区的单碱基突变(错义突变或同义突变)激活邻近的隐蔽裂解信号,影响IVS正常位点的剪接,产生异常的mRNA。如东南亚常见HbE,是一种轻型的β地贫,其原因是当β链26位密码子发生G→A错义突变时,其相邻的裂解信号被激活,生成异常mRNA,产生HbE(图4-16)。
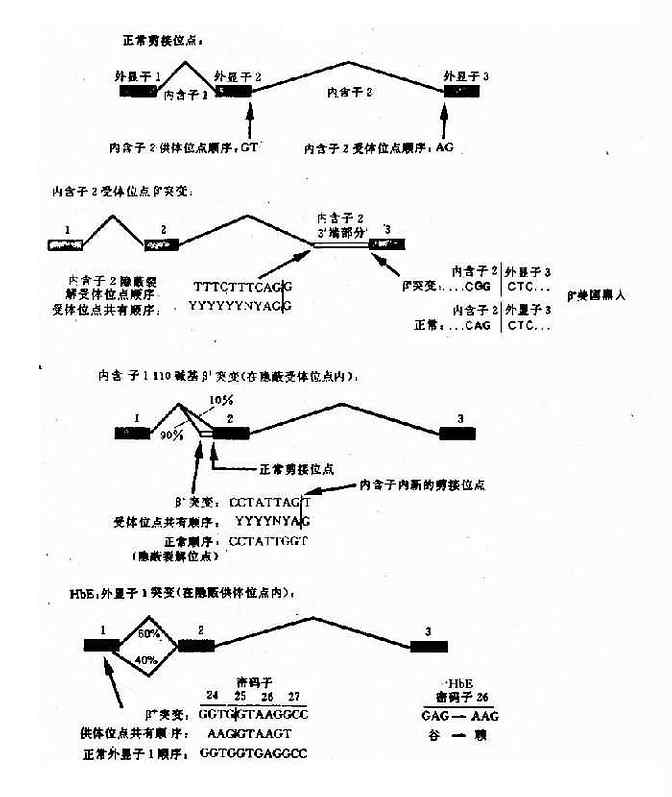
图4-16 干扰正常β珠蛋白剪接的突变举例
HbE:密码子26(G-→A)
谷→赖
GAG→AAG(QAJ)(错义突变)
β+(HbA 60%) βE(HbE 40%)
3)类β基因缺失:①按类β珠蛋白基因簇缺失长短大致可分为4种,即β0、δβ、γδ地中海贫血及遗传性胎儿血红蛋白持续增多症;②单纯由于β0基因缺失引起的β地中海贫血罕见;③融合基因,如HbLeproe,是类β基因缺失7kb导致δβ融合基因,形成β0地贫。
| 关于“医学遗传学/血红蛋白病的分类和分子基础”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |