医院药学/药品生产管理规范
| 医学电子书 >> 《医院药学》 >> 医院制剂业务管理 >> 药品生产管理规范 |
| 医院药学 |
|
|
GMP(good practice the manfacture and quality control of drugs或称Good Manufacturing)译为“优良的生产”或译为“药品生产管理规范”。它开始是由美国坦普尔大学6名教授编写制定的,1963年由美国国会第一次颁布布成为法令。在1969年,第二十二届世界卫生大会的决议要求所有会员国执行《药品生产管理规范》。后来,在第二十四届世界卫生大会上,又作出决定,现成世界卫生组织的理事长继续研究上述文件中的规定,是否切实可行,并向第二十五届世界卫生大会作出报告。据此,世界卫生组织现令其专门设置的专家委员会负责,研究各会员国提出的意见,并草拟一个修正方案。这一方案于1974年分发全体会员国,随后收到约四十个国家的答复。经专家委员会再次讨论最后于1975年第十一月提出了修改后的《药品生产管理规范》,1977年第二十八届世界卫生大会讨论通过,确定为世界卫生组织的法规。目前世界上已有一百多个国家执行GMP。有一些先进国家还制订了本国的GMP,GMP分药品生产的厂房、设备,人员、原材料、工艺规程、生产记录、生产控制、监控方法、产品包装、销售和稳定性等章节。它的基本指导思想是,用全面质量管理,保证药品的安全性、有效性、稳定性和品质优良,它要求药品生产的质量管理不限于分析、化验、检查表格,车间检验和出厂检验等,而是涉及整个生产全过程的进行质量监控措施。
(一)GMP的特点
1、它强调药品生产和质量管理的法律责任。只要生产药品就要向卫生行政部门登记,就要按GMP要求,接受卫生行政部门的监督。
2、对影响产品质量的因素有严格要求,强调生产人员的业务能力、技术水平和教育。
3、强调生产全过程和全面性的质量管理。
4、强调防检结合,以防为主。
5、广泛应用数理统计方法。对抽样和统计的控制限度作了具体规定,以保证样本的代表性,防止造成无根据的结论。
6、重视为用户服务。要求建立销售档案,保证药品的稳定性,制订有效期,建立申诉档案。
多年的GMP实施结果证明,GMP可以把人为事故降低到最小限度,从根本上保证药品质量。
(二)GMP的种类
现行的GMP有三种:一是国际的,如WHO提出的GMP;北欧七国自由贸易聪明制定的PIC。二是国家级的,如美国、加拿大、澳大利、日本等都有本国的GMP,它由国家制定,由卫生行政部门监督实施。三是医药工业界自定的GMP。
WHO推荐的 GMP是药品生产和质量管理通用的指南,可使用国家间的投资,合资经营的药品进出口贸易便利,使相互之间的监督检查有统一的标准。我国制定GMP后,也便于药品进出口。
(三)国外GMP概要
1、美国坦普尔大学提出的GMP 坦普尔大学6位教授指出的全面质量管理方案分十五章。第一章定义,第二章成药,第三章厂房,第四章设备,第五章人员,第六章原材料,第七章工艺流程与生产记录,第八章生产和监控过程,第九章产品容器,第十章包装与贴签,第十一章化验室管理,第十三章稳定性,第十四章制订有效期,第十五章申诉档案。
在第一章中,不仅确定了产品种类和名称,还提出了物料和文件流动的模式示意图,见图12-1。
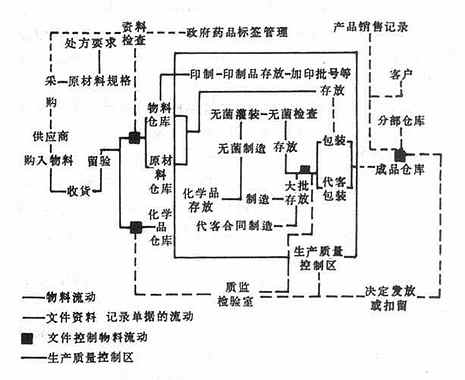
图12-1 美国GMP物料及文件流动模式图
2、世界卫生组织公布的GMP 世界卫生组织(WHO)于1979年在第二十八届世界卫生大会上通过的GMP共分十三章。
第一章概论。其中提到,为保证消费者能获得高质量的药品,在生产中实行全面管理是十分重要的。生产药品时决不允许出现任意的操作方式。优良的制度应被视作通用的准则。
第二章定义。其中提高药品、生产、批、批号、留验、质量控制、半成品、起始原料等定义。
第三章人员。其中提到负责指导药品生产和质量控制的专家,应具备国家法令规定的专门训练和经验的资格。他们受的教育应包括:化学;化学工程学;微生物学;药剂学和技术;药理学和毒理学;生物学和组织学;其它有关科学。除了上面说的专家以外,还应有一定数量的,经过技术训练的人员,来进行生产和质量控制。
第四章厂房建筑。所有的药品均应在适合条件的厂房建筑里进行生产、加工、包装、贴签和试验。其中提到厂房建筑时,要注意与相邻厂房生产操作的相互影响,要有适当的工作场所,防止混药、防止污染等问题。
第五章设备。生产用设备必须适合指定用途,便于进行清扫,污染降至最低限度,无菌灌注用的设备,每隔适当阶段用微生物学方法加以核查。生产用的量、衡设备,每隔一定阶段加以校正和核查。
第六章卫生。所有厂房均应清洁,要求打扫的区域规定每次打扫的期限,需要采取的打扫办法,指定专人负责清洁卫生工作。在工作区附近,应该有足够清洁的、通风良好的盥洗设施,包括洗手装置和更衣室等。
第七章起始原料。药品生产各个阶段所用的一切起始原料都应有所库存,并应保存关于供应单位、来源、收货日期、分析日期、质量监督部门批准使用日期和用于生产的日期等。凡是被接受或批准的起始原料均应有妥善和明显的标志。所有无法使用的起始原料均应有明显标志,并应忙地予以处理或退回供应单位。
第八章生产操作。所有生产操作和控制都应在专家指导下进行。操作要特别注意:清洁工作、设备容器、污染和混杂的预防、灭菌操作的特殊要求、抗生素类生产的特殊要求等;对生产人员要求:不能患有传染病骸身体裸露、表面患有开放性疾病,工作服、鞋帽不能脏污等;对与生产程序有关文件的制订、生产批次记录等。其物料文件流程要求如图12-1。
第九章标签和包装。标签和包装材料,包括说明书在内,必须按不同的产品分别储存和处理,只有经过授权的人员方能接触这种材料。标签和包装材料应包括书籍发放数量、包装和贴签后应该计数量。所有已经编号而未用的标签应全部销毁。
第十章质量监控制度。每一个生产单位都应有一个质量监督部门,在有适宜资格的专家指导下进行工作,直接对总管理负责,而独立于其它的部门。质量监督部门必须控制所有的起始原料,监查生产操作中有关质量部分,并对药品质量和稳定性实行监督。质量监督部门必须有一个专用的实验室。
第十一章自检。为了能严格的与所有生产程序及规定的控制要求相符合,制药企业可考虑指定一个专家或一批专家,负责经常地对它的整个生产及控制操作进行定期检查。
第十二章发送记录。对于已经制成的每一批药品,都应保存关于发送情况的合适记录,以便于必要时能迅速而全部地将这批药品撤回。
第十三章关于不良反应的报告和控诉。关于用药后引起损害或不良反应的报告,必须送交适当的负责人审阅,有关药品质量的控诉,必须彻底地进行查究。如果证明这些控诉确有根据,应尽可能迅速地采取适当措施。已经采取的措施必须记录下来,并与原来的控诉一起归档。
| 关于“医院药学/药品生产管理规范”的留言: | |
|
目前暂无留言 | |
| 添加留言 | |